click here for video of breathing lung
click here for enhanced view of an ex vivo lung
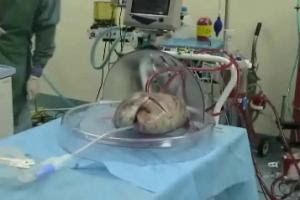
"After the donor lungs are removed from a deceased donor and taken to the hospital, they are carefully transferred to a protective, transparent bubble-like chamber that the Toronto team developed in collaboration with Vitrolife, a company specializing in developing lung preservation solutions.
"To avoid injuring the lungs, a series of precise steps are followed when connecting them to a circuit composed of a pump, ventilator and filters through which flow oxygen, nutrients and a special solution The temperature is incrementally increased until it reaches 37 degrees Celsius over about 30 minutes, and ventilation of the lungs is begun during that time." I was honored to participate in the news conference in Toronto announcing this medical breakthrough that will ultimately save many more lives by getting more patients off the waiting lists and giving them the 'Gift of Life'.
Congratulations to the surgeon who saved my life with a single lung transplant, Dr. Shaf Keshavjee and his Lung Transplant Team for this achievement that will have a global impact. Merv.
December 19, 2008
TORONTO —
University Health NetworkFor the first time in the world, transplant surgeons at Toronto General Hospital, University Health Network used a new technique to repair an injured donor lung that was unsuitable for transplant, and then successfully transplanted it into a patient.
The use of this technique could significantly expand the lung donor organ pool and improve outcomes after transplantation.
In their ground-breaking research, a team of lung transplant surgeons led by Dr. Shaf Keshavjee in the Lung Transplant Program at Toronto General Hospital (TGH) developed an “ex vivo” or outside the body technique capable of continuously perfusing or pumping a bloodless solution containing oxygen, proteins and nutrients into injured donor lungs.
This technique, the Toronto XVIVO Lung Perfusion System, allows the surgeons the opportunity to assess and treat injured donor lungs, while they are outside the body, to make them suitable for transplantation.
Unlike current cooling lung preservation techniques which inhibit cell metabolism and the possibility of any active repair processes prior to transplantation, the Toronto technique maintains donor lungs at a normal body temperature of 37 degrees Celsius, allowing for future organ repair and gene and cell therapy strategies to be used on them.
Although lung perfusion systems have been used in Sweden and England, these systems were blood-based, short-term assessment strategies which were not capable of long-term maintenance or techniques to repair or recondition the lungs.
The Toronto System was used on donor lungs for Andy Dykstra, 56, who received his transplant on December 5, 2008. He had been waiting for a transplant since July 30, 2008, and was told of the clinical trial testing the new system on December 4, 2008.
Recalling his difficulty breathing when walking to the front door in his home, Andy said that he waited only two seconds before deciding to be the first patient to receive the reconditioned lungs, which would not have been suitable for transplant without undergoing perfusion by the Toronto System.
“When I was given this chance, the hair on my arms stood up, I was so excited. I knew it was right. I just had to go for it,” he said, with his wife Chris standing by his side and nodding for emphasis.
Andy is part of an ongoing clinical trial which uses the novel Toronto strategy to identify donor lungs which do not meet current transplant criteria, repair them, and then transplant them into patients. To date, four patients in total have received lungs treated using this technique, and all have done well.
But Andy was the first patient who received lungs which did not meet standard transplant criteria and which could not have been used if they had not been repaired first by the Toronto System. (The three others received donor lungs which met transplant criteria and which were further improved by the Toronto System.)
All TGH patients waiting for a lung transplant are eligible to be part of this clinical trial, and the lung transplant team will assess all those who are interested.
“We are extremely pleased that Andy is doing so well,” said Dr. Keshavjee who is Director of the Lung Transplant Program at TGH, Senior Scientist, The McEwen Centre for Regenerative Medicine, Director, Latner Thoracic Research Laboratories, and Professor and Chair, Division of Thoracic Surgery, University of Toronto, adding that Andy was able to breathe without any mechanical assistance just four days after the transplant and was discharged from hospital 12 days after the procedure.
“This achievement was the result of years of research and pre-clinical planning by a large team of researchers, surgeons, physicians, nurses and other specialists. It means that many more donor lungs which we could not have used before can now potentially be used safely, and it sets the stage for more sophisticated molecular and cellular repair techniques to be applied in the Toronto XVIVO Perfusion System so that transplant outcomes can be further improved. The potential exists to immunologically pre-prepare the organ before it even sees the recipient’s immune system.”
He explained that currently only about 15%-20% of donor lungs are acceptable for transplantation since lungs are susceptible to injuries during the brain-death process or from intensive care unit-related lung complications. These numbers can easily be doubled with this technique to treat and improve donor lungs.
Dr. Marcelo Cypel, a transplant surgical fellow at TGH, echoed Dr. Keshavjee’s sentiments. “This new technique heralds the beginning of a new era in transplantation since it has allowed us to progress from preserving donor lungs to actually being able to repair some of the injury before transplantation. And we have done this using a unique strategy on donor lungs outside the body.”
After the donor lungs are removed from a deceased donor and taken to the hospital, they are carefully transferred to a protective, transparent bubble-like chamber that the Toronto team developed in collaboration with Vitrolife, a company specializing in developing lung preservation solutions.
To avoid injuring the lungs, a series of precise steps are followed when connecting them to a circuit composed of a pump, ventilator and filters through which flow oxygen, nutrients and a special solution The temperature is incrementally increased until it reaches 37 degrees Celsius over about 30 minutes, and ventilation of the lungs is begun during that time.
Lung function is evaluated regularly on key indicators, such as how easily the lungs can exchange oxygen, airway pressure and lung compliance.
Previously published research by Drs. Keshavjee and Cypel on this system (December 2008 issue of the Journal of Heart and Lung Transplantation) has shown that lungs can be safely kept on this circuit for 12 hours in order to assess, maintain and treat them before successfully transplanting them.
Currently, about 80 patients are waiting for either a lung or heart-lung transplant in Ontario. About 20% of those on the wait list will die before they receive a lung transplant.
In Canada, the number of people waiting for a lung transplant has doubled in the past 10 years, with 252 Canadians waiting to receive a lung transplant in 2006, compared to 119 in 1997. Two hundred and ninety-nine (299) Canadians died while waiting for a lung transplant between 1997 and 2006.
The clinical trial is funded by Vitrolife Inc. and is approved by the Research Ethics Board of University Health Network.
A Canadian Institute for Health Information report this week showed that 299 people died while waiting for a lung transplant between 1997 and 2006.
However, the number of lung transplants increased to 171 in 2006 from 93 in 1997, while the three-year survival rate has improved to 80 per cent in 2003 from 60 per cent in 1997, CIHI said.
“You Have the Power to Save Lives – Sign Your Donor Card & Tell Your Loved Ones of Your Decision”
Register to be a donor in Ontario or Download Donor Cards from Trillium Gift of Life Network
For other Canadian provinces click here
In the United States, be sure to find out how to register in your state at ShareYourLife.org or Download Donor Cards from OrganDonor.Gov
Your generosity can save up to eight lives with heart, kidneys, liver, lungs, pancreas and small intestine transplants. One tissue donor can help up to 100 other people by donating skin, corneas, bone, tendon, ligaments and heart valves









